Introduction
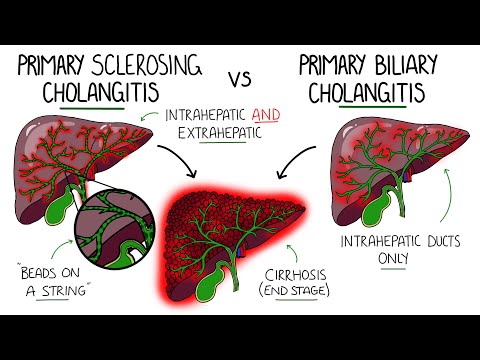
Primary Biliary Cholangitis (PBC) is a chronic autoimmune disorder characterized by progressive destruction of intrahepatic bile ducts, leading to cholestasis, liver damage, and potential cirrhosis, necessitating comprehensive management and therapeutic approaches.
Causes and Risk Factors
PBC etiology remains unclear, but genetic predisposition, environmental triggers, and immune system dysregulation contribute to its development, predominantly affecting middle-aged women, with associations also seen in familial cases and certain geographical regions.
Autoimmune Nature of PBC
Primary Biliary Cholangitis is characterized by a breakdown in immune tolerance, resulting in a multifaceted autoimmune response targeting the intrahepatic bile ducts. This response is mediated by autoreactive T cells and B cells, which orchestrate a complex interplay of cellular and humoral immunity.
Elevated levels of autoantibodies, including antimitochondrial antibodies (AMA) and anti-nuclear antibodies (ANA), are commonly observed in PBC patients, further supporting the autoimmune nature of the disease. The presence of these autoantibodies often precedes clinical symptoms, suggesting a prolonged subclinical phase of autoimmunity.
The precise mechanisms underlying this autoimmune response remain unclear, but it is believed that a combination of genetic predisposition, environmental triggers, and molecular mimicry contribute to the development of PBC. Elucidating the immunological pathways involved in PBC is crucial for the development of targeted therapeutic strategies and improved disease management.
A thorough understanding of the autoimmune processes driving PBC will facilitate the identification of potential biomarkers for early diagnosis, monitoring, and prediction of treatment outcomes, ultimately enhancing patient care and quality of life.
Liver Damage and Chronic Inflammation
Persistent autoimmune targeting of the intrahepatic bile ducts in Primary Biliary Cholangitis results in chronic inflammation, leading to progressive liver damage and scarring. Activated immune cells, including T cells and macrophages, infiltrate the liver parenchyma, releasing pro-inflammatory cytokines and chemokines that perpetuate tissue damage.
The ongoing inflammatory response causes the destruction of bile ducts, disrupting normal bile flow and leading to the accumulation of toxic bile acids within the liver. This, in turn, triggers a cascade of cellular and molecular events that contribute to hepatocellular injury and fibrogenesis.
Chronic inflammation and liver damage also promote the activation of hepatic stellate cells, which undergo phenotypic transformation into myofibroblast-like cells. These cells play a key role in the deposition of extracellular matrix proteins, driving the progression of liver fibrosis and, ultimately, cirrhosis.
The complex interplay between immune-mediated damage, bile acid toxicity, and fibrogenesis underlies the pathogenesis of PBC, highlighting the need for a multidisciplinary approach to disease management and therapeutic intervention.
Understanding the mechanisms of liver damage and chronic inflammation is crucial for the development of novel treatments aimed at halting disease progression and improving clinical outcomes in PBC patients.
Symptoms and Diagnosis
Primary Biliary Cholangitis often presents with nonspecific symptoms, including fatigue, pruritus, and jaundice, necessitating a comprehensive diagnostic approach incorporating clinical evaluation, laboratory testing, and imaging studies to confirm the diagnosis and assess disease severity.
Jaundice Symptoms and Other Clinical Manifestations
Primary Biliary Cholangitis often presents with jaundice symptoms, characterized by yellowing of the skin and sclera due to elevated bilirubin levels. Additional clinical manifestations may include⁚
- Pruritus⁚ intense itching sensations, particularly at night, which can significantly impact quality of life.
- Fatigue⁚ persistent and debilitating exhaustion, affecting daily activities and overall well-being.
- Xanthelasmata⁚ small, yellowish patches or nodules on the skin, particularly around the eyes.
- Xanthoma⁚ larger, yellowish lesions on the skin, often associated with hypercholesterolemia.
These symptoms can vary in severity and may be intermittent, making diagnosis and disease monitoring challenging. Furthermore, some patients may experience additional symptoms, such as abdominal pain, nausea, and vomiting, which can further compromise quality of life. A comprehensive clinical evaluation is essential to identify and manage these symptoms effectively, ensuring optimal patient outcomes.
Diagnostic Tests and Procedures
Accurate diagnosis of Primary Biliary Cholangitis requires a combination of serological tests, imaging studies, and histopathological evaluation. Key diagnostic tests and procedures include⁚
- Antimitochondrial antibody (AMA) testing⁚ detection of AMA-M2 is highly specific for PBC.
- Liver function tests (LFTs)⁚ assessment of alkaline phosphatase, gamma-glutamyl transferase, and bilirubin levels.
- Ultrasound and magnetic resonance cholangiopancreatography (MRCP)⁚ visualization of bile ducts and liver morphology.
- Liver biopsy⁚ histopathological evaluation of liver tissue to assess disease severity and stage.
A diagnosis of PBC is typically made when two or more of the following criteria are met⁚ presence of AMA, abnormal LFTs, and characteristic histopathological features. In some cases, additional testing, such as computed tomography (CT) or endoscopic retrograde cholangiopancreatography (ERCP), may be necessary to rule out other conditions or evaluate disease complications.
Treatment and Management
The primary goals of treatment for Primary Biliary Cholangitis are to slow disease progression, manage symptoms, and prevent complications, utilizing a combination of pharmacological interventions, lifestyle modifications, and close monitoring of liver function.
Ursodeoxycholic Acid Therapy
Ursodeoxycholic acid (UDCA) is the first-line treatment for Primary Biliary Cholangitis, with a recommended dose of 13-15 mg/kg/day. UDCA has been shown to slow disease progression, improve liver function tests, and reduce the risk of liver transplantation and death.
The mechanism of action of UDCA involves modification of bile acid composition, reduction of toxic bile acid levels, and stabilization of cell membranes. This leads to decreased apoptosis and inflammation in the liver, ultimately slowing disease progression.
Clinical trials have consistently demonstrated the efficacy and safety of UDCA in patients with Primary Biliary Cholangitis. Response to treatment is typically assessed through monitoring of liver function tests and clinical evaluation. Adverse effects are rare, but may include gastrointestinal disturbances, pruritus, and hepatotoxicity.
Early initiation of UDCA therapy is crucial to maximize treatment benefits. Patients should be regularly monitored to assess response to treatment and adjust the dosage as needed. UDCA therapy has significantly improved the prognosis and quality of life for patients with Primary Biliary Cholangitis, making it an essential component of disease management.
Cholestasis Management and Fatigue Treatment Options
Management of cholestasis in Primary Biliary Cholangitis involves alleviating symptoms and preventing complications. Pruritus is a common symptom, often treated with antipruritic agents, bile acid sequestrants, and opioid antagonists.
Fatigue is a debilitating symptom affecting quality of life in patients with Primary Biliary Cholangitis. Treatment options include behavioral modifications, such as regular exercise and cognitive behavioral therapy, to improve sleep quality and reduce fatigue.
Medications like modafinil and armodafinil have shown promise in reducing fatigue in some patients. However, these agents should be used judiciously, considering potential side effects and interactions with other medications.
In addition to pharmacological interventions, lifestyle modifications, including a balanced diet and stress management, can help alleviate fatigue and improve overall well-being. A multidisciplinary approach, involving healthcare providers, nutritionists, and mental health professionals, is essential for effective cholestasis management and fatigue treatment in patients with Primary Biliary Cholangitis.
Regular monitoring and individualized treatment plans are crucial to address the complex and multifaceted nature of fatigue in Primary Biliary Cholangitis, ensuring optimal management and improved quality of life for affected patients.
Complications and Prognosis
Untreated Primary Biliary Cholangitis can lead to severe complications, including cirrhosis, liver failure, and increased risk of hepatocellular carcinoma, emphasizing the importance of prompt diagnosis, effective management, and regular monitoring to optimize prognosis and patient outcomes.
Cirrhosis Risk Factors and Liver Transplantation
Cirrhosis is a significant complication of Primary Biliary Cholangitis, with risk factors including prolonged disease duration, advanced age at diagnosis, and inadequate treatment response. Histological features, such as interface hepatitis and ductular reaction, also contribute to cirrhosis development.
Liver transplantation remains the definitive treatment for end-stage liver disease secondary to Primary Biliary Cholangitis. Patients with decompensated cirrhosis, characterized by jaundice, ascites, or variceal bleeding, should be referred for transplant evaluation in a timely manner.
Recurrence of Primary Biliary Cholangitis after liver transplantation has been reported, underscoring the need for ongoing monitoring and potential reintroduction of ursodeoxycholic acid therapy. Collaborative management between hepatologists and transplant specialists is crucial to optimize patient outcomes and minimize post-transplant complications.
Early recognition of cirrhosis risk factors and consideration of liver transplantation can significantly improve survival rates and quality of life for patients with advanced Primary Biliary Cholangitis. Regular follow-up and multidisciplinary care are essential to address the complex needs of these patients.
Long-term Outlook and Disease Monitoring
The long-term outlook for patients with Primary Biliary Cholangitis is variable, with some individuals experiencing slow disease progression and others rapidly advancing to cirrhosis. Regular monitoring is essential to assess treatment response, detect potential complications, and adjust management strategies accordingly.
Disease monitoring should include routine liver biochemistry tests, such as alkaline phosphatase and bilirubin levels, as well as periodic assessment of liver stiffness by elastography or biopsy. This enables healthcare providers to track disease progression and adjust ursodeoxycholic acid dosing or add alternative therapies as needed.
Patient education and engagement are critical components of long-term disease management. Individuals with Primary Biliary Cholangitis should be aware of the importance of adherence to treatment regimens, regular follow-up appointments, and prompt reporting of new symptoms or concerns.
By fostering a collaborative relationship between patients and healthcare providers, optimal disease control can be achieved, and quality of life improved. Furthermore, ongoing research into novel therapeutic agents and biomarkers holds promise for enhancing treatment outcomes and personalizing care for patients with Primary Biliary Cholangitis.
Primary Biliary Cholangitis is a complex and multifaceted autoimmune disorder that necessitates comprehensive management and individualized care. Through ongoing research and advances in therapeutic strategies, patients with this condition can experience improved outcomes and enhanced quality of life.
A multidisciplinary approach, incorporating hepatology, gastroenterology, and primary care expertise, is essential for delivering optimal care and addressing the diverse needs of individuals with Primary Biliary Cholangitis.
Future directions in the field of Primary Biliary Cholangitis research include the development of novel therapeutic agents, exploration of biomarkers for disease diagnosis and monitoring, and elucidation of the underlying pathophysiological mechanisms driving disease progression.
Ultimately, continued collaboration and knowledge-sharing among healthcare professionals, researchers, and patients will facilitate advancements in the understanding and management of Primary Biliary Cholangitis, fostering improved treatment outcomes and better patient care.
By prioritizing patient-centered care, promoting awareness, and supporting ongoing research endeavors, we can work towards a future where individuals with Primary Biliary Cholangitis receive the most effective and compassionate care possible, enabling them to thrive and maintain an optimal quality of life.
The section on liver damage and chronic inflammation effectively highlights the consequences of untreated PBC. Nevertheless, I believe it would be beneficial to include more information on preventive measures or lifestyle changes that can help manage the condition.
I appreciate how this article delves into the complexities of PBC
This article demonstrates a thorough understanding of PBC
This article provides an excellent overview of Primary Biliary Cholangitis (PBC), covering its definition, causes, risk factors, autoimmune nature, liver damage, and chronic inflammation. The author
Overall, this article provides a solid foundation for understanding Primary Biliary Cholangitis. One area for improvement could be adding more visual aids or diagrams to illustrate key concepts and enhance reader comprehension.